Language

 动物实验
动物实验
 z-bo
z-bo
 2020-08-27
2020-08-27
 980
980
Can simple genetic risk assessments identify complex diseases? The development of gene expression profiles for acute myeloid leukemia suggests that they can do it, and they may improve the prognosis of this disease.
Researchers such as
Ng reported in an online article published in the journal Nature that a tool that can improve the prognosis of patients with a subtype of acute leukemia. First, researchers identify cell populations that can express important characteristics, which are collectively called stemness, which enables cells to induce and maintain leukemia. This allows the authors to determine the expression patterns of stemness-related genes and use them as the basis for a risk scoring system. This research work shows how gene expression profiles can be used as a reliable prognosis for complex diseases.
Acute myeloid leukemia (AML) is characterized by a large number of chromosomal and molecular aberrations. This means that AML patients with widely differing prognosis have many subgroups. These groups are called risk groups, and they are used to determine which consolidation treatments should be given after initial therapy (induction therapy). For example, for patients with leukemia diagnosed as the most dangerous, stem cell transplantation can be chosen, but because these transplants often have fatal side effects, it is not the best choice for some patients. Therefore, the improvement of risk assessment is necessary, not only for the formulation of consolidation treatment strategies, but also for the selection of different types of induction therapy (it is expected to become available in the future).
Gene expression profiling may help achieve these improvements. The method of Ng and colleagues relies on the recognition of dry gene expression patterns, which is a good proof of the application of gene expression profiling. In normal tissues, stemness enables stem cells to renew themselves to maintain the long-term differentiation process of normal cells. In the hematopoietic system, normal hematopoietic stem cells (HSC) are the source of circulating blood cells and bone marrow. Hematopoietic stem cells can express CD34 protein on their surface, but not CD38 protein, so they have CD34+CD38-immune phenotype. Leukemia stem cells (LSCs) have stem properties similar to hematopoietic stem cells, but they can express different patterns of cell surface proteins: they can be CD34+CD38- cells (this may be derived from hematopoietic stem cells), but they can also have CD34+ CD38+ , CD34 − CD38 +, CD34 − CD38 − immunophenotype. It has been demonstrated in animal models that these different CD34/CD38 cell subsets have different abilities to induce leukemia.
Through tremendous efforts, Ng et al. isolated 227 cell components defined by CD34/CD38 from 78 patients with acute myeloid leukemia and injected these cell components into mice (Figure 1). They confirmed that there are differences in the ability of these cell components to initiate leukemia: Leukemia can form all the cell components obtained from a patient, or some, or even not at all. Then, the author compared the gene expression of the original cell components, no matter which CD34/CD38 cell components, these original cell components are caused by gene expression in the cell components without leukemia induced leukemia. This allowed them to determine gene expression patterns, which are directly related to the ability of cells in mice to form leukemia.
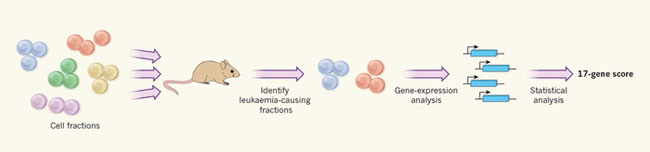
Figure 1: The 17-gene score for assessing the risk of acute leukemia.
Ng et al. obtained cell samples from patients with acute myeloid leukemia, and divided them into different cell components based on the CD34 and CD38 proteins expressed on the cell surface. The researchers transplanted these cellular components into mice and determined which components caused leukemia and which components did not cause leukemia. The authors then compared the gene expression patterns of pathogenic and non-pathogenic cell components to identify candidate genes related to tumor formation. This information is used to guide the statistical analysis of gene expression data collected in clinical studies of acute myeloid leukemia patients. Statistical analysis can calculate the patient's score based on the expression of 17 genes. This score provides a reliable method for evaluating the prognosis of patients.
Ng and colleagues first identified 104 differential genes based on at least two-fold difference in gene expression between the leukemia initiating cell component and the non-leukemia initiating cell component. Then the author obtained a large amount of gene expression data from clinical studies of 495 patients with acute myeloid leukemia, and found that 89 genes out of 104 differential genes exist in this set of data. The cells in this study were not classified into components, but the gene expression patterns displayed were similar to those observed by researchers such as Ng in the initiating cell components of leukemia.
Next, the authors used a statistical method to correlate gene expression with clinical results to study these 89 genes and 43 genes that are highly expressed in the initial cell fraction of leukemia. This allowed them to determine the optimal set of 17 genes, which expression has high predictive value for the adverse clinical outcome of patients in the disease subgroup. The authors confirmed this finding in other AML groups and discovered a scoring system based on their genome (known as LSC17). Compared with other gene expression pattern systems in patients with acute myeloid leukemia, LSC17 can achieve a better prognosis. In fact, Ng and colleagues found that the previously reported genetic characteristics of AML patients cannot be used as an independent prognostic factor when testing other AML groups.
Researchers such as
Ng also found that the gene expression pattern associated with AML stemness is independent of the chromosomal and molecular aberrations used to assess patient risk, indicating that cell stemness is a prognostic factor in addition to the previously determined risk group. Finally, the author developed a test that can quickly generate gene expression data, which can form a rapid-based (24-48 hours) patient prognosis prediction.
As the author pointed out, to evaluate whether the prognostic value of the LSC17 score has nothing to do with the prognostic value of the mutation, a large amount of data analysis in clinical studies is required. At the same time, extensive information about the mutation status of leukemia cells and the LSC17 score in these clinical studies are both available. acquired. The clinical benefit of the LSC17 score is a must be evaluated, because the prognostic value does not always produce a meaningful clinical advantage. In addition, a small population of leukemia cells with similar genetic makeup (clones) can exist in disease diagnosis, survival treatment, and proliferation leading to recurrence (which occurs after additional mutations). Only time will tell whether gene expression profiles can explain these clone-defined cellular components and predict the recurrence of related diseases.
The prognosis of leukemia patients at the time of diagnosis is only a part of the whole process of disease prognosis. Once treatment is started, many factors such as treatment dependence, changes in the concentration of drugs used to alleviate side effects, and differences in the concentration of drugs in the patient's plasma may partly mask the effects of prognostic diagnostic parameters, such as gene expression patterns. Consideration of post-treatment parameters such as predictable (mild) sequelae (the persistence of a small number of white blood cells during the course of the patient’s symptom relief) has completely changed the current status of risk assessment for AML patients. Combining the characteristics of the cells at the time of diagnosis, the non-cellular patient-specific factors during treatment, the frequency and characteristics of cells maintained after treatment, and the changes in immune parameters may provide a more complete prognosis than currently predicted. This will enable more personalized induction and consolidation treatments to be applied. The study by Ng and colleagues may be a great advancement in this type of evaluation, especially in the diagnosis phase.
